La alfa manosidosis es una enfermedad rara de almacenamiento lisosomal provocada por mutaciones que afectan a la enzima lisosomal alfa manosidasa. Esta enzima es una exoglicosidasa, que separa los residuos de manosa de los oligosacáridos. Si se daña la enzima alfa manosidasa, se bloquea la degradación de las glucoproteínas y se produce una acumulación progresiva de oligosacáridos ricos en manosa en todos los tejidos, lo que conlleva a un deterioro de las funciones celulares y finalmente a la apoptosis.1

Este deterioro de las funciones celulares tiene un impacto posterior en los órganos, con manifestaciones típicas como deformaciones del esqueleto, rasgos faciales toscos, pérdida de audición, discapacidades cognitivas, defectos inmunitarios y deterioro del sistema nervioso central.2 Se desconoce la prevalencia exacta de la alfa manosidosis. Sin embargo, según varios informes de diferentes países, se calcula que se da aproximadamente en uno de cada millón de bebés que nacen en el mundo.3 La enfermedad suele ser diagnosticada y tratada con un enfoque multidisciplinar en el que se incluyen pediatras, traumatólogos, oftalmólogos, neurólogos, inmunólogos, neurocirujanos y fisioterapeutas.4 La alfa manosidosis es un trastorno crónico, y debe sospecharse su presencia en pacientes con discapacidad cognitiva, alteraciones en el esqueleto (articulaciones inflamadas, escoliosis, etc.), pérdida de audición e infecciones recurrentes. Aunque los niños con la enfermedad suelen nacer aparentemente normales, su estado se deteriora con la edad. La alfa manosidosis puede afectar a la calidad de vida de los pacientes de muchas maneras, incluida su capacidad de vivir de forma independiente, socializar o encontrar un empleo.5 6
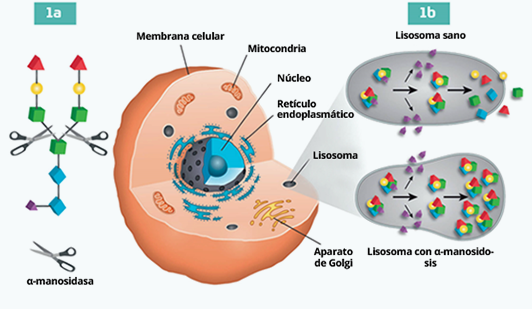
Mecanismo de
la enfermedad
El objetivo de la información contenida en este sitio web es únicamente dar a conocer temas relacionados con la enfermedad alfa-manosidosis. Esta información no debesustituir el consejo de su médico de cabecera o de otro profesional sanitario. Si tiene dudas, póngase en contacto con su médico. Este sitio web ha sido producido porChiesi Pharmaceuticals. El sitio web ha sido desarrollado de conformidad con los estándares industriales y jurídicos para aportar información a profesionales sanitarios y alpúblico en general sobre temas relacionados con la enfermedad alfa-manosidosis. Chiesi Pharmaceuticals se esfuerza al máximo por incluir información precisa yactualizada. No obstante, la información contenida en este sitio web no es exhaustiva.




