Encuentre la respuesta a preguntas frecuentes sobre la alfa manosidosis.
Las preguntas se organizan por tema para facilitar la búsqueda.
Pase el cursor por encima de cada pregunta para ver la respuesta que necesita.
La alfa manosidosis es una enfermedad hereditaria rara que puede provocar que niños y adultos sufran deformidades óseas, rasgos faciales toscos, pérdida de audición, retraso mental, problemas con el sistema inmunitario, trastornos psiquiátricos y problemas de comportamiento.1 Cuando el gen que proporciona las instrucciones para producir la alfa manosidasa no funciona correctamente, la enzima no se fabrica, lo que causa la alfa manosidosis.2 Esta enzima trabaja en los lisosomas, que son los orgánulos que digieren y reciclan los materiales en la célula. Dentro de los lisosomas, la enzima ayuda a descomponer largas cadenas de moléculas de azúcar (oligosacáridos). Los oligosacáridos se emplean para formar los huesos, cartílagos, piel, tendones y muchos otros tejidos del organismo.3 4 En las personas con alfa manosidosis, los azúcares parcialmente descompuestos se almacenan en el organismo y se acumulan con el tiempo. Esto produce daños en las células.5Es posible que los bebés apenas muestren indicios de la enfermedad, pero conforme van resultando dañadas más células por la acumulación de oligosacáridos, van apareciendo los síntomas.6
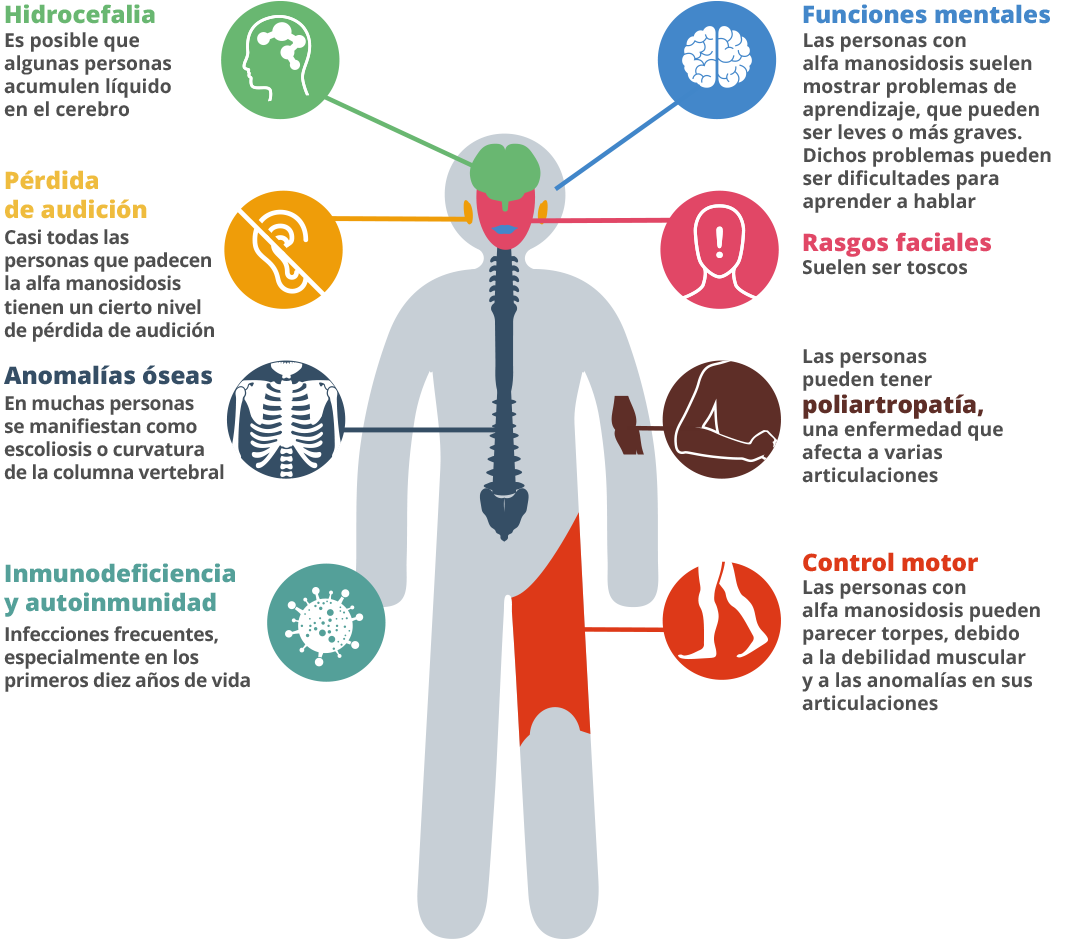
Los síntomas de la alfa manosidosis, al igual que los de otras enfermedades similares pueden ser muy variables.7 Se trata de una enfermedad progresiva, que se manifiesta de diferentes maneras a lo largo del tiempo.8 Debido a su amplio número de síntomas, y dado que cada persona presenta su propio cuadro, el diagnóstico puede tardar mucho tiempo.9 En la primera década de vida, los niños afectados pueden tener infecciones frecuentes, problemas de audición, rasgos faciales diferenciados y retrasos en el desarrollo.10 11 Los padres pueden notar cierta debilidad muscular en los niños.12 Puede aparecer una característica extraña, como un pie equino varo, una cabeza demasiado grande, una apariencia inusual o quizás una espalda encorvada. Los niños pueden tener problemas de atención y dificultades auditivas.13 14 Un síntoma puede ser un caso aislado, pero la combinación de síntomas al mismo tiempo no es una coincidencia. Tenga en cuenta todos los síntomas y pida a su médico de familia que le derive a un especialista en enfermedades metabólicas. Los adultos de 20 a 40 años pueden experimentar problemas óseos y dificultades de movimiento, como problemas articulares, inflamación, andar inestable y debilidad muscular. Al final, los pacientes pueden depender de una silla de ruedas, ya que dejan de poder andar por sí solos.15 Es posible que tengan problemas de comportamiento o psiquiátricos, que pueden presentarse como episodios de confusión, a veces acompañados de ansiedad, alucinaciones o depresión.16 Es difícil que lleven una vida independiente, y es posible que los pacientes con alfa manosidosis sufran aislamiento social.17 18 El pronóstico a largo plazo de la enfermedad es desfavorable.19
Se han propuesto tres subtipos clínicos:37
Sin embargo, dada la variedad de mutaciones que se han documentado, el amplio espectro y gravedad de los síntomas y la ausencia de vínculos entre mutaciones concretas y subtipos, a nivel clínico la enfermedad se considera como un continuo de leve a grave.38 39
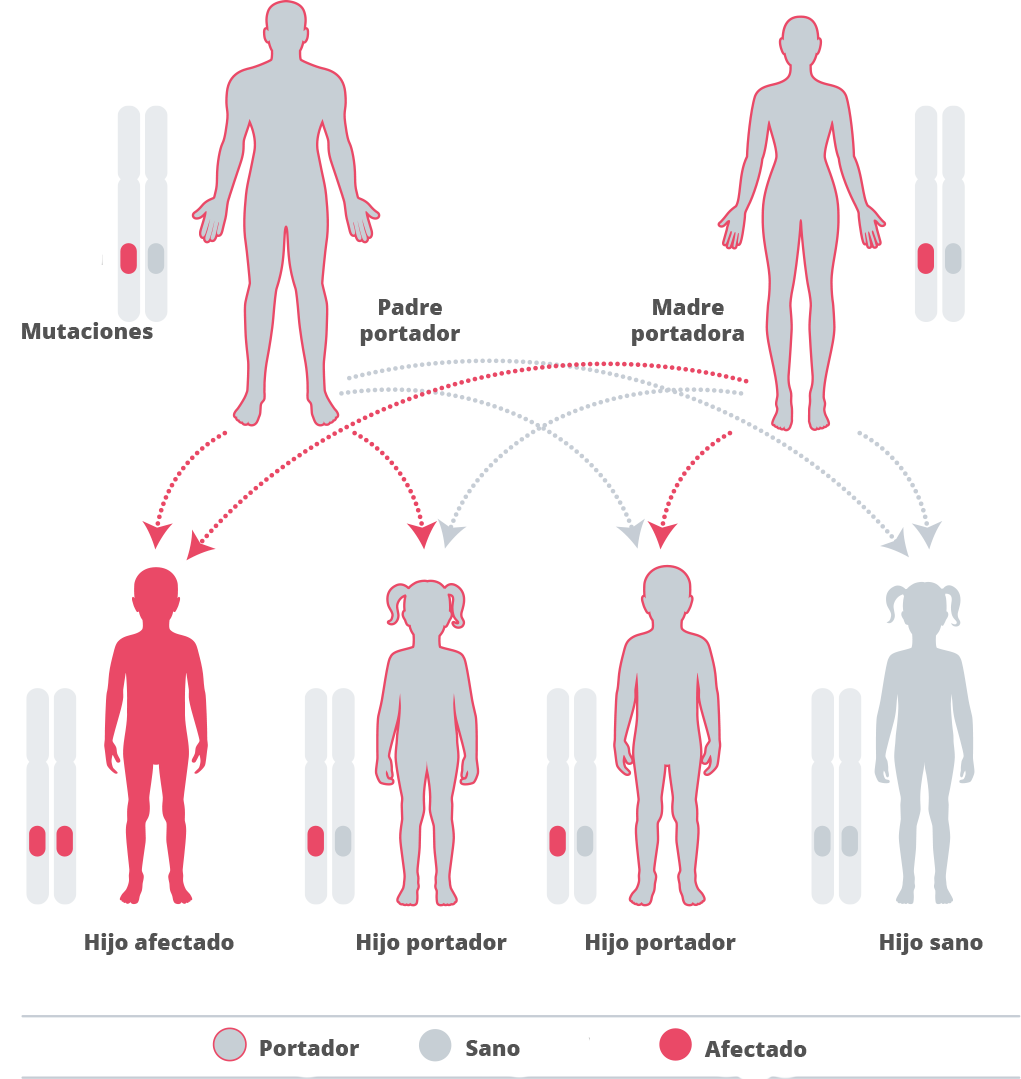
La alfa manosidosis es hereditaria, lo que significa que depende de la familia.40 Los genes se heredan de nuestros padres. Heredamos una copia de cada gen de cada uno de nuestros padres. Algunos de los genes que heredamos son recesivos, lo que significa que tienen que heredarse dos copias del gen para que tenga algún efecto en nuestro desarrollo.41 42 La alfa manosidosis está causada por un gen recesivo.43 En los embarazos en los que ambos progenitores sean portadores heterocigotos para alguna mutación en el gen MAN2B1 causante de la enfermedad, presentan una probabilidad del 25 % de que nazca un bebé afectado, una probabilidad del 50 % de que nazca un bebé no afectado pero que sea portador y una probabilidad del 25 % de que nazca un bebé no afectado que no sea portador.44
Dado que la enfermedad es tan rara, la posibilidad de tener una pareja que también sea portadora es escasa, a menos que los individuos sean de la misma familia.45 Si una pareja tiene un hijo con la enfermedad, se recomienda asesoramiento genético para comprender las probabilidades de que la enfermedad vuelva a estar presente en otros hijos.46
No se conoce con exactitud la prevalencia de la alfa manosidosis. Sin embargo, según varios informes de diferentes países, se da aproximadamente en uno de cada millón de bebés que nacen en el mundo.47
Su médico puede emplear varias pruebas sencillas para diagnosticar la alfa manosidosis, como, por ejemplo:
Existe una comunidad de soporte ahí fuera que está deseando proporcionar fuentes de información, comprensión y consejo. Hay muchísimas organizaciones y grupos de apoyo a unos pocos clics de distancia. Organizaciones de pacientes:
 es el grupo de apoyo internacional de la alfa manosidosis es el grupo de apoyo para familias en Nueva Zelanda..
es el grupo de apoyo internacional de la alfa manosidosis es el grupo de apoyo para familias en Nueva Zelanda..