Trouvez ici des réponses aux questions courantes concernant l’alpha-mannosidose Les questions sont classées par thème afin de faciliter la recherche. Cliquez sur chaque question pour trouver la réponse que vous cherchez.
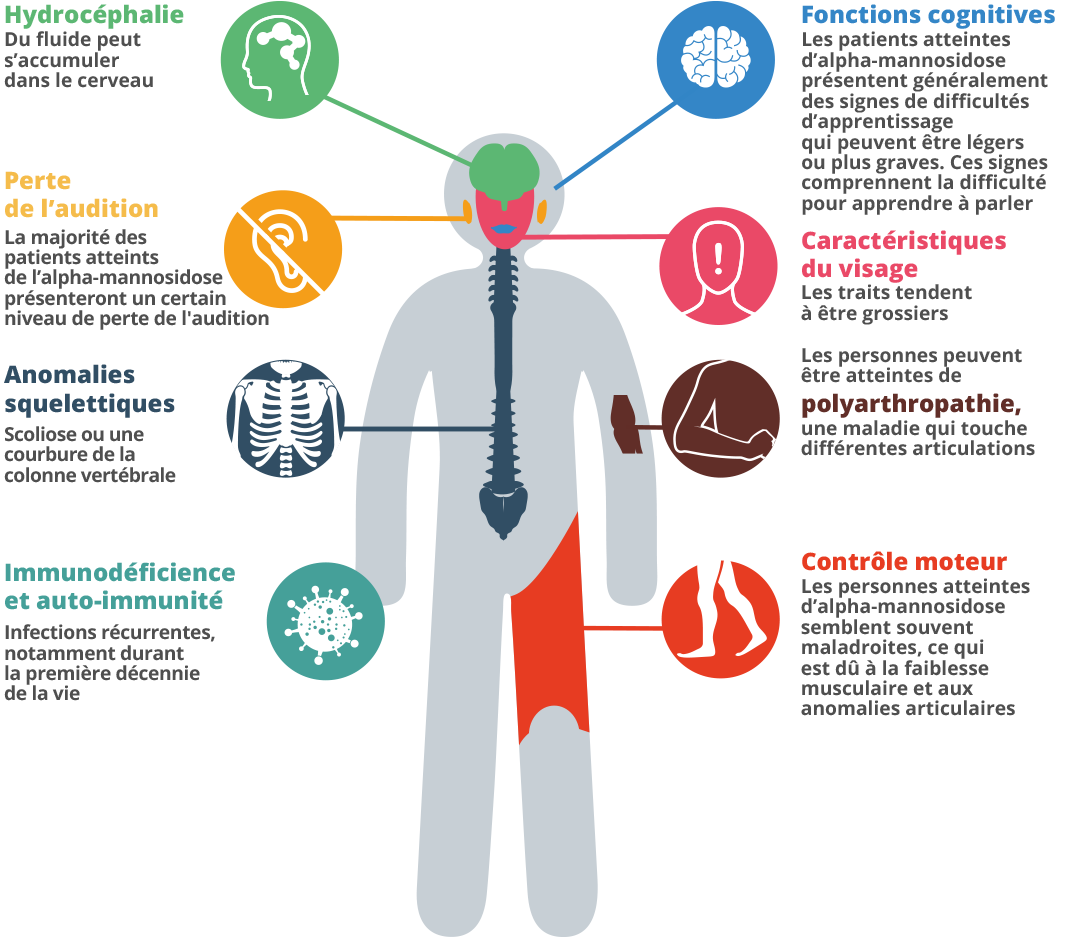
L’alpha-mannosidose est une maladie héréditaire rare qui peut engendrer, chez l’enfant et l’adulte, des malformations squelettiques et faciales, la perte de l’audition, un retard mental, des troubles psychiatriques, des problèmes du système immunitaire et des problèmes de comportement.1 Lorsque le gène qui fournit les instructions pour fabriquer l’alpha-mannosidose ne fonctionne pas correctement, l’enzyme n’est pas produite correctement, ce qui en résulte l’alpha-mannosidose.2 Cette enzyme est présente dans les lysosomes, qui sont les compartiments qui digèrent et recyclent le matériel dans la cellule. Dans les lysosomes, l’enzyme aide à cliver les longues chaines de molécules de sucres (oligosaccharides). Les oligosaccharides sont utilisés dans la constitution des os, du cartilage, de la peau, des tendons et de nombreux autres tissus du corps.3 4 Chez les personnes atteintes d’alpha-mannosidose, les sucres partiellement dégradés sont conservés dans le corps et s’accumulent avec le temps, ce qui engendre des dommages croissants des cellules.5 Chez les bébés les signes de la maladie peuvent être faibles, mais de plus en plus de cellules sont endommagées par l’accumulation d’oligosaccharides et les symptômes commencent à se manifester.6
Les symptômes de l’alpha-mannosidose, comme d’autres troubles apparentés, peuvent être très variables.7 Il s’agit d’une maladie évolutive, qui se manifeste de différentes manières dans le temps.8 Le nombre de symptômes étant très important, et chaque individu ayant des symptômes qui lui sont propres, le diagnostic peut être long.9 Durant les dix premières années de vie, un enfant atteint de cette maladie peut être sujet à des infections fréquentes, des problèmes d’audition, des traits du visage distinctifs et un retard du développement.10 11 Les parents peuvent remarquer que les muscles de leur enfant sont faibles.12 Parmi les effets peuvent également figurer le pied bot, une grande tête, une apparence inhabituelle, parfois un dos courbé. Un enfant peut avoir des troubles de l’attention et de l’audition.13 14 Un symptôme peut être ponctuel, mais la combinaison de plusieurs symptômes en même temps n’est pas une coïncidence. Considérez tous les symptômes, et consultez un spécialiste du métabolisme en demandant conseil à votre médecin traitant. Lorsqu’il a 20 et 30 ans, un adulte est sujet à des problèmes osseux et des difficultés de mouvement, comme des problèmes des articulations, de déglutition, une démarche instable et une faiblesse des muscles. Enfin, les patients arrivent à dépendre d’un fauteuil roulant, car ils ne marchent plus tout seuls.15 Ils peuvent avoir des problèmes du comportement ou psychiatriques, qui peuvent se traduire par des épisodes de confusion, parfois accompagnés d’anxiété, de dépression ou d’hallucinations.16 La vie indépendante devient difficile, et les patient atteints d’alpha-mannosidose peuvent devenir socialement isolés.17 18 Les prévisions à long terme de cette maladie sont faibles.19
Trois sous-types cliniques ont été proposés :37
Toutefois, au vu du grand nombre de mutations qui ont été relevées, de la multitude et de la gravité des symptômes, de l’absence de liens entre des mutations particulières et un sous-type clinique, cette maladie est considérée comme ayant une progression de modérée à grave.38 39
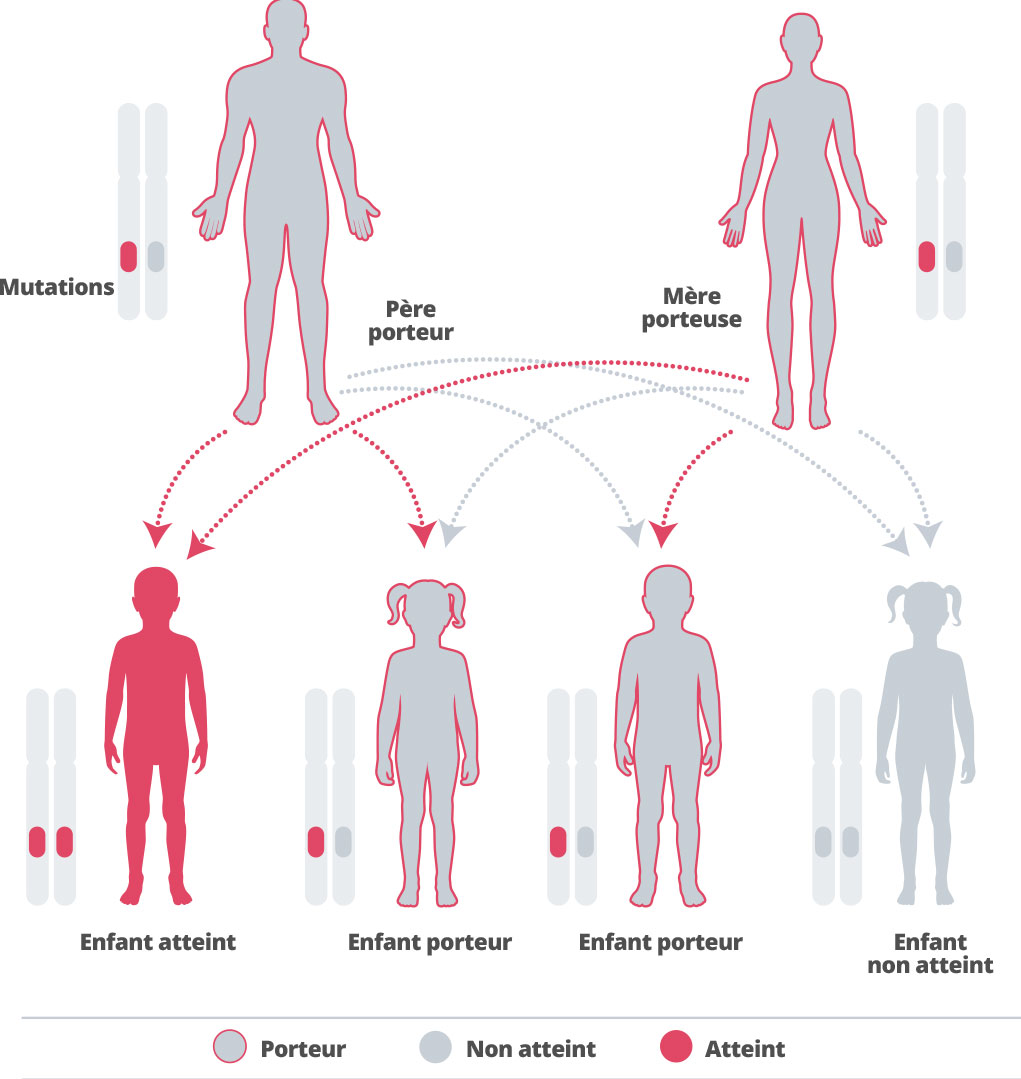
L’alpha-mannosidose est héréditaire, ce qui veut dire qu’elle se trouve dans les gènes40 dont nous héritons de nos parents. Nous héritons un exemplaire de chacun des gènes de chacun de nos parents. Certains gènes hérités sont « récessifs », ce qui signifie que les deux exemplaires de ce gène doivent être hérités pour qu’il ait un effet sur notre développement.41 42 L’alpha-mannosidose est provoquée par un gène récessif.43Pour un couple où les 2 conjoints sont hétérozygotes avec la mutation du gène MAN2B1 (responsable de la maldie), le risque d’avoir un enfant atteint pour chaque grossesse est de 25%. Le risque d’avoir un enfant non atteint porteur est de 50% et le risque d’avoir un enfant non atteint non porteur est de 25%.44 Cette maladie étant très rare, la possibilité d’avoir un partenaire qui est également porteur est faible, sauf si les individus font partie de la même famille.45 Si un couple a déjà un enfant atteint de la maladie, il est conseillé de consulter un généticien afin de comprendre les probabilités que la maladie soit également transmise aux enfants futurs.46
La prévalence de l’alpha-mannosidose n’est pas connue de manière précise. Toutefois, d’après un certain nombre de rapports provenant de différents pays, il est estimé qu’elle touche environ 1/500000 à 1/1 million nouveau-né dans le monde.47
Votre médecin peut utiliser différents tests très simples pour diagnostiquer l’alpha-mannosidose, comme par exemple :
Il existe une communauté de soutien qui est heureuse de fournir des informations, sa compréhension et ses conseils. Les associations et groupes de soutien ne sont qu’à quelques clics. Associations de patients :
Le diagnostic d’alpha-mannosidose peut avoir un impact émotionnel considérable sur les patients et le personnel soignant. L’accès à des soins primaires et spécialisés, ainsi qu’aux services sociaux, doivent être assurés, et une aide nutritionnelle et psychologique doit être évaluée périodiquement.52 Les soins médicaux doivent être proactifs, c’est-à-dire que les médecins doivent prévoir de limiter les conséquences de la maladie et toute complication pour les patients.53 La chirurgie conjointe ou orthopédique peut être prise en considération.54 Pour de plus amples informations quant aux options possibles pour la gestion, n’hésitez pas à en parler avec votre médecin.
Les informations figurant sur ce site web sont uniquement destinées à fournir des connaissances sur les thématiques de santé liées à l’alpha-mannosidose. Ces informations ne doivent pas remplacer les conseils de votre médecin ou d’un autre professionnel de santé. En cas de doute, veuillez contacter votre médecin pour obtenir des conseils. Ce site web a été réalisé par Chiesi Pharmaceuticals. Il a été développé conformément aux normes industrielles et juridiques afin de fournir des informations aux professionnels de santé et au grand public sur des thématiques de santé liées à l’alpha-mannosidose. Chiesi Pharmaceuticals fait tout son possible pour inclure des informations précises et actuelles. Toutefois, les informations fournies sur ce site ne sont pas exhaustives.