Trova le risposte alle domande più comuni sull'Alfa-mannosidosi.
Troverai le domande organizzate per temi in modo da facilitare la ricerca.
Sposta il cursore su ogni domanda per trovare la risposta che stai cercando.
L’Alfa-mannosidosi è una malattia rara ed ereditaria che può colpire adulti e bambini con deformità dello scheletro, deformità facciali, perdita dell’udito, ritardi mentali, problemi del sistema immunitario, disturbi psichiatrici e problemi comportamentali.1 Quando il gene che fornisce istruzioni per creare l’alfa-mannosidasi non funziona correttamente, l’enzima non viene prodotto nel modo giusto e causa l’Alfa-mannosidosi.2 Questo enzima lavora nei lisosomi, che sono compartimenti in grado di digerire e riciclare materiali nelle cellule. Nei lisosomi l’enzima aiuta a disintegrare le molecole di zucchero a catena lunga (oligosaccaridi). Gli oligosaccaridi vengono usati dalle cellule nella formazione delle ossa, della cartilagine, della pelle, dei tendini e di molti altri tessuti del corpo.2,3 Nelle persone con l’Alfa-mannosidosi, gli zuccheri parzialmente disintegrati rimangono immagazzinati nel corpo e si accumulano nel tempo, danneggiando progressivamente le cellule.2 I bambini inizialmente possono mostrare pochi segni della malattia, ma i sintomi iniziano a manifestarsi con il passare del tempo quando le cellule vengono sempre più danneggiate dall’accumulo di oligosaccaridi.3
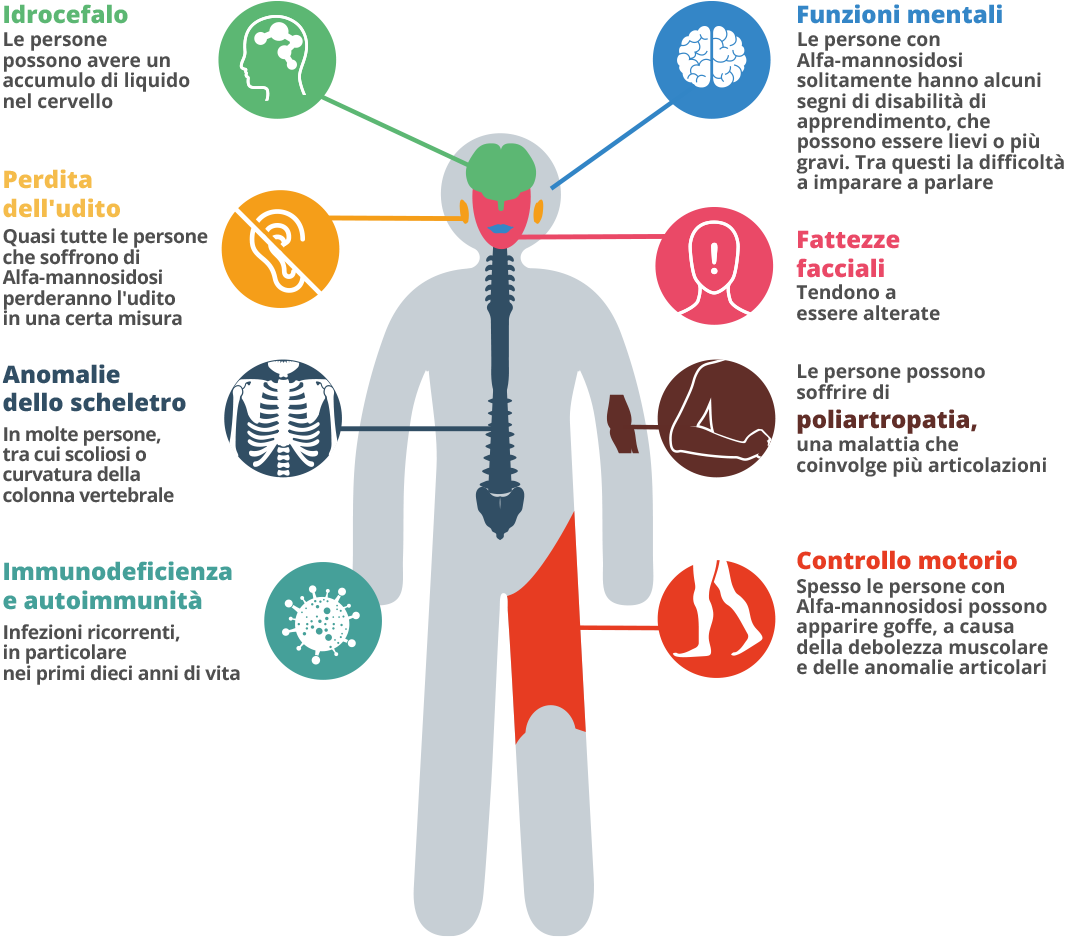
I sintomi dell’Alfa-mannosidosi, come altre malattie rare, possono essere molto diversi.1 È una malattia progressiva che si manifesta in modi diversi lungo un determinato periodo di tempo.4 Data la vasta gamma di sintomi e il fatto che si manifestino in ogni individuo in uno schema unico, la diagnosi può richiedere molto tempo.4 Durante i primi dieci anni di vita, il bambino con questo disturbo può soffrire di frequenti infezioni, problemi all’udito, caratteristiche facciali anomale e di un ritardo nello sviluppo.1,5 I genitori possono notare un’eventuale debolezza muscolare del figlio.4 Potrebbe esserci una caratteristica inusuale come per esempio un piede equino, la testa larga, caratteristiche facciali e corporee alterate e in alcuni casi la schiena curva. Il bambino può avere problemi d’attenzione e difficoltà d’udito.3,4 Un solo sintomo potrebbe essere un’eccezione, ma la combinazione di più sintomi allo stesso tempo non è una coincidenza. Considera tutti i sintomi e rivolgiti a uno specialista delle malattie del metabolismo, chiedendo al tuo medico di famiglia di consigliarti. Tra i 20 e i 30 anni, un adulto potrebbe avere problemi e difficoltà nel movimento, problemi alle articolazioni, gonfiori, andatura instabile e debolezza muscolare. I pazienti possono infine diventare dipendenti da una sedia a rotelle perché ormai incapaci di camminare da soli.1 Potrebbero manifestarsi problemi psichiatrici e comportamentali che possono presentarsi come episodi di confusione, a volte accompagnati da ansia, depressione o allucinazioni.1 L’indipendenza nella vita quotidiana potrebbe essere difficile e i malati di Alfa-mannosidosi potrebbero venire isolati dalla vita sociale.1,6 La prognosi a lungo termine per questa malattia al momento è limitata.1
Sono stati proposti tre sottotipi clinici:1
Tuttavia, vista la varietà di mutazioni che sono state documentate, la vasta gamma e gravità di sintomi e l’assenza di un collegamento tra specifiche mutazioni e sottotipi clinici, la malattia è considerata clinicamente come un continuum da lieve a grave.5,6
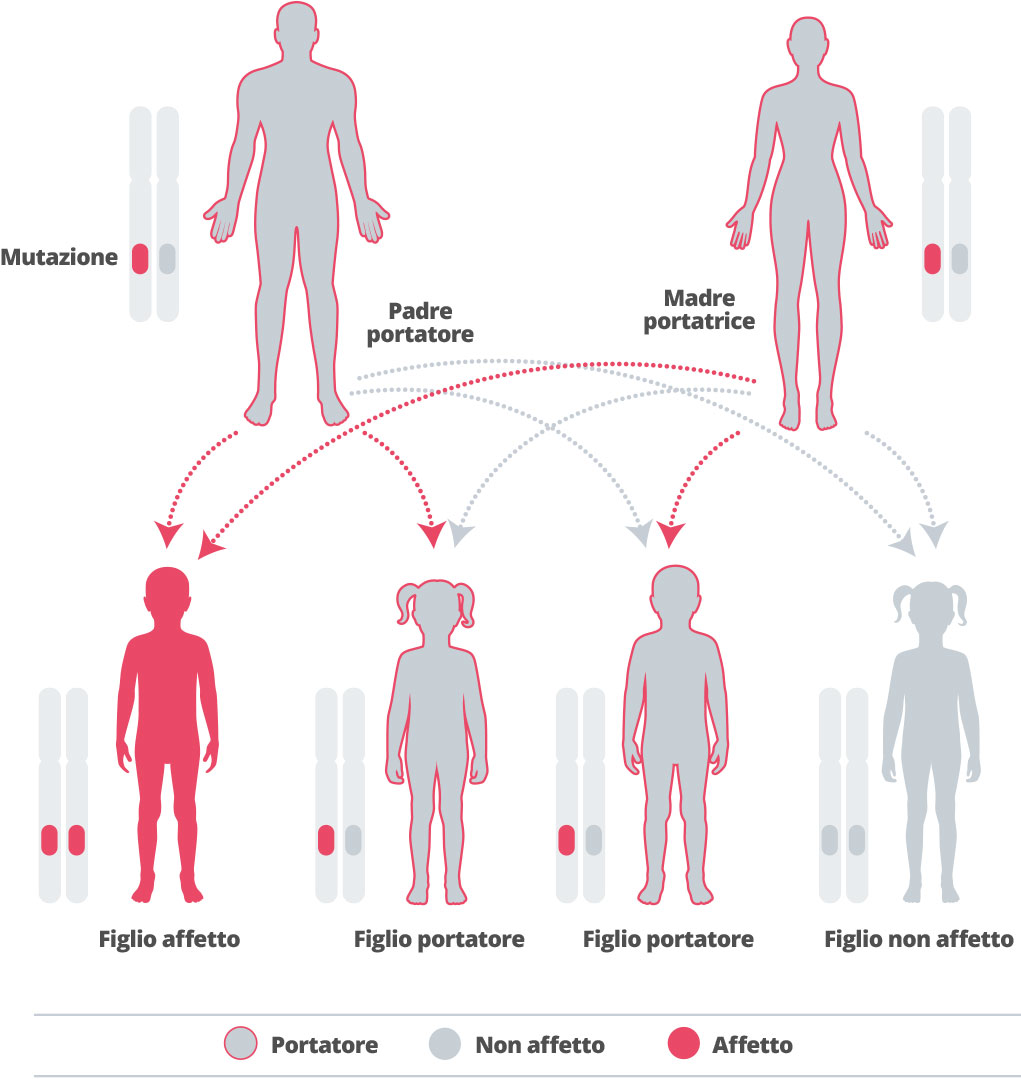
L’Alfa-mannosidosi è ereditaria, quindi si trasmette in famiglia di generazione in generazione.1 I geni si ereditano dai genitori. Ereditiamo una copia di ogni gene da ognuno dei genitori. Alcuni geni che ereditiamo sono “recessivi” – che significa che dovremmo ereditare entrambe le copie di quel gene perché esso abbia un effetto sul nostro sviluppo.7,8 L’Alfa-mannosidosi è causata da un gene recessivo.1 Ciascuna gravidanza di una coppia in cui entrambi i partner sono eterozigoti per una mutazione del gene MAN2B1 che causa la malattia presenta una probabilità del 25% di avere un figlio che possa soffrire della malattia, una probabilità del 50% di avere un figlio non affetto ma portatore sano e una probabilità del 25% di avere un figlio non affetto e non portatore. 7
Dal momento che la malattia è così rara, la possibilità di avere un partner che è a sua volta portatore è bassa, a meno che gli individui non siano della stessa famiglia.3 Se una coppia ha già avuto un bambino con la malattia, si raccomanda una consulenza genetica, per capire le possibilità che la malattia possa comparire nella futura prole.3
La prevalenza dell’Alfa-mannosidosi non è ancora nota in termini esatti. Tuttavia, una serie di rapporti da diversi paesi stimano che la malattia si manifesti approssimativamente in un bambino su un milione di nascite in tutto il mondo.5
Il tuo dottore può usare una serie di test molto semplici per diagnosticare l’Alfa-mannosidosi come:
Esiste una comunità di sostegno pronta a dare informazioni, solidarietà e consigli. Varie associazioni e gruppi di sostegno sono a distanza di pochi click da te. Associazioni di pazienti:
La diagnosi dell’Alfa-mannosidosi può avere un considerevole impatto emozionale sui pazienti e su chi si prende cura di loro. L’accesso alle cure primarie e specialistiche e i servizi sociali devono essere garantiti su base continua.9 Le cure cliniche dovrebbero essere preventive e i medici devono pianificare le cure per limitare le conseguenze della malattia ed eventuali complicanze per i pazienti.1 Si possono considerare interventi chirurgici alle articolazioni o altri interventi ortopedici.1 Per ulteriori informazioni sulle opzioni di gestione della malattia è bene discuterne con il proprio medico.
Le informazioni che si trovano su questo sito sono volte soltanto a fornire conoscenze riguardanti questioni di salute sulla malattia dell’Alfa-mannosidosi. Queste informazioni non devono essere usate al posto di un consiglio del proprio medico o di un professionista della salute. In caso di dubbi contattare il proprio medico. Questo sito è stato prodotto da Chiesi Farmaceutici. Il sito è stato sviluppato in accordo con gli standard legali e di settore per fornire informazioni ai professionisti della salute e al pubblico in generale sulle tematiche della patologia Alfa-mannosidosi. Chiesi Farmaceutici compie il massimo sforzo per inserire informazioni accurate e aggiornate. Tuttavia, le informazioni qui fornite non sono esaustive.


