Encontre suas respostas para perguntas comuns sobre Alfamanosidose. As perguntas são organizadas por tópico para facilitar a pesquisa. Passe o mouse sobre cada pergunta para encontrar a resposta de que você precisa.
Alfamanosidose é uma doença rara, hereditária que pode fazer com que crianças e adultos sofram com alterações esqueléticas, características faciais grosseiras, perda de audição, deficiências cognitivas, problemas no sistema imunológico (o que pode significar, por exemplo, infecções com maior frequência), problemas de saúde mental e comportamentais. 1
Essa enzima trabalha nos lisossomos, que são compartimentos que digerem e reciclam materiais na célula. Nos lisossomos, a enzima ajuda a quebrar as moléculas de açúcar chamadas oligossacarídeos. São moléculas de açúcar de cadeia longa usadas na construção de ossos, cartilagens, pele, tendões e muitos outros tecidos do corpo. 3,4
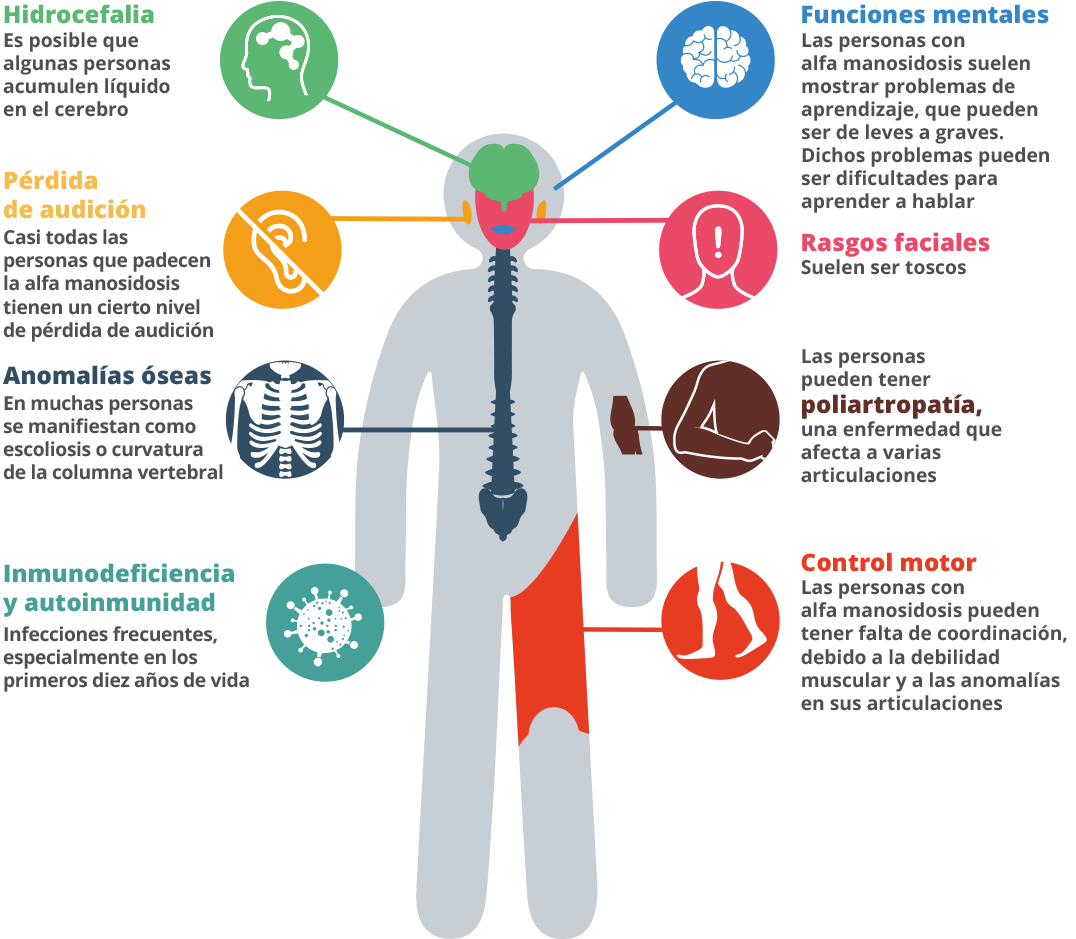
Os sintomas da Alfamanosidose, como outras doenças relacionadas, podem ser muito variáveis.7
É uma doença progressiva, que se manifesta de várias maneiras ao longo do tempo.8
Por apresentar uma ampla gama de sintomas e como cada indivíduo experimentará seu próprio padrão único de sintomas, o diagnóstico pode levar muito tempo.9
Durante a primeira década de vida, uma criança com a doença pode apresentar infecções frequentes, problemas auditivos, características faciais distintas e atraso no desenvolvimento.10 11
Os pais podem notar que seus filhos podem ter alguma fraqueza muscular.12 Pode haver uma característica incomum, como um pé torto, uma cabeça grande, aparência incomum, talvez as costas curvadas. Uma criança pode ter problemas de atenção e dificuldade para ouvir.13 14 Um sintoma pode ser único, mas a combinação de sintomas ao mesmo tempo não é coincidência. Considere todos os sintomas e busque o encaminhamento para um especialista em metabolismo, pedindo ao seu médico de família para encaminhá-lo.
Na casa dos 20 e 30 anos, um adulto pode ter problemas ósseos e dificuldades de movimento, como problemas nas articulações, inchaço, marcha instável e fraqueza muscular. Em última análise, os pacientes podem se tornar dependentes de uma cadeira de rodas, pois não podem mais andar por conta própria.15
Pode haver problemas comportamentais ou psiquiátricos, que podem se apresentar como episódios de confusão, às vezes com ansiedade, depressão ou alucinações.16
Uma vida independente será difícil, e os pacientes com Alfamanosidose podem se tornar socialmente isolados.17 18 A previsão de longo prazo para a condição é ruim.19
Foram sugeridos pelo menos três tipos clínicos de Alfamanosidose:37
No entanto, dada a variedade de mutações que foram documentadas, a ampla gama e gravidade dos sintomas e nenhuma ligação entre mutações específicas e sintomatologia, a doença é considerada clinicamente como um continuum, de leve a grave.38,39
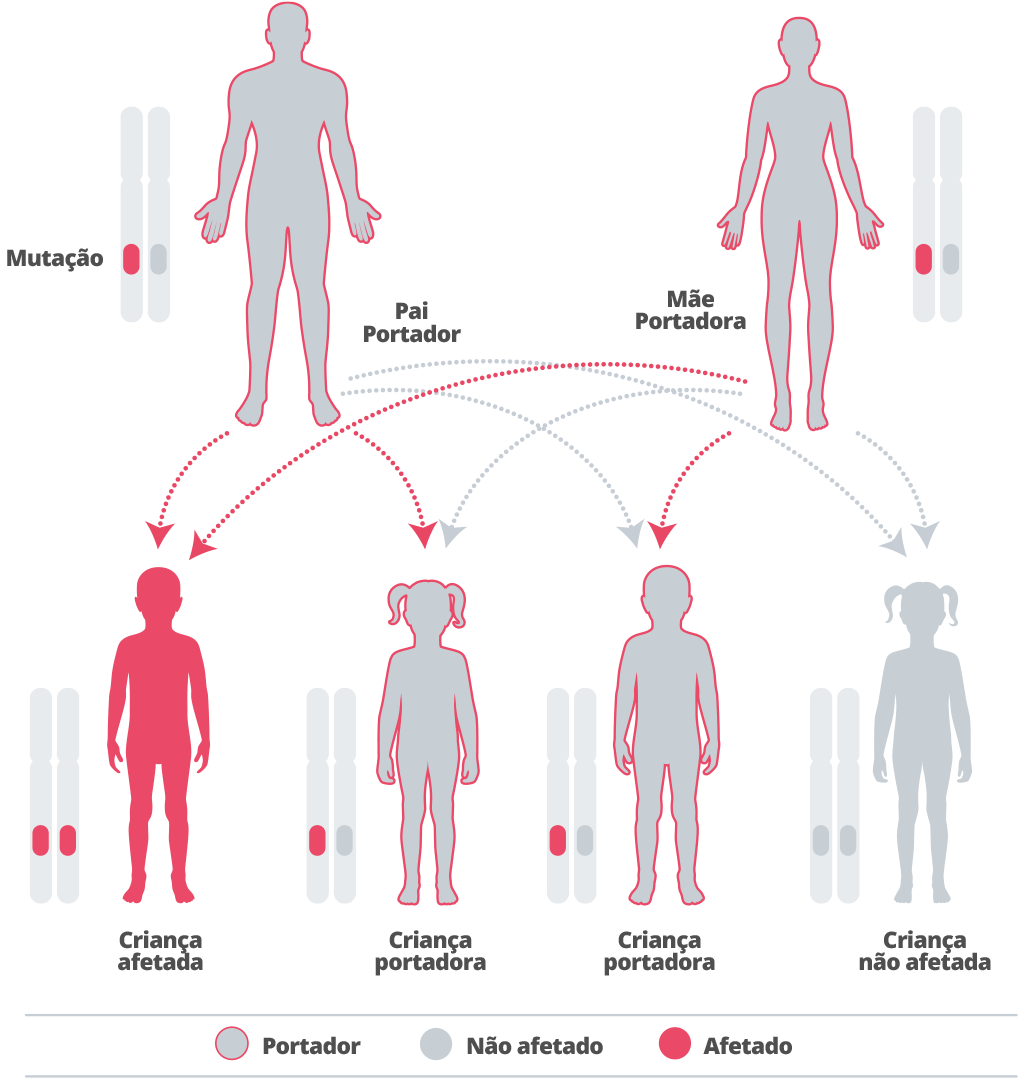
Alfamanosidose é hereditária, o que significa que ocorre em famílias.40
Os genes são herdados de nossos pais. Herdamos uma cópia de cada gene de cada pai. Alguns genes que herdamos são “recessivos”, o que significa que ambas as cópias desse gene precisam ser herdadas para que tenha algum efeito em nosso desenvolvimento.41 42
Alfamanosidose é causada por um gene recessivo.43Cada gravidez de um casal em que ambos os parceiros são heterozigotos para uma mutação causadora da doença no gene MAN2B1 tem 25% de chance de ter um filho afetado, 50% de chance de ter um filho não afetado que seja portador e 25% de chance de ter um filho não afetado que não seja portador.44
Como a doença é rara, a chance de ter um parceiro que seja outro portador é pequena, a menos que os indivíduos sejam da mesma família.45 Se um casal já tem um filho com a doença, recomenda-se aconselhamento genético, para entender as chances da doença também ocorrendo em descendentes futuros.46
A prevalência de Alfamanosidose não é conhecida exatamente. No entanto, vários relatórios de diferentes países estimam que ocorra em aproximadamente um em cada milhão de bebês nascidos em todo o mundo.47
Seu médico pode usar uma série de testes muito simples para diagnosticar Alfamanosidose, incluindo:
Existe uma comunidade de suporte por aí que está ansiosa por fornecer informações, compaixão e conselhos. Quando você está se sentindo mal (e haverá momentos), várias organizações e grupos de suporte estão a apenas alguns cliques de distância para fornecer suporte.
Organizações de pacientes:
O diagnóstico de Alfamanosidose pode ter um impacto emocional considerável nos pacientes e cuidadores. O acesso aos cuidados primários e especializados e aos serviços sociais deve ser fornecido, e o suporte nutricional e psicológico deve ser avaliado em uma base contínua.52 O atendimento clínico deve ser proativo, com médicos planejando para limitar as consequências da doença e quaisquer complicações para os pacientes.53 Para obter mais informações sobre as opções de tratamento, converse com seu médico.
As informações contidas neste site destinam-se apenas a fornecer conhecimento dos conceitos de saúde da doença Alfamanosidose. Estas informações não devem ser usadas no lugar de aconselhamento do seu médico ou outro profissional de saúde. Em caso de dúvida, entre em contato com seu médico para obter orientação. Este site foi produzido pela Chiesi Pharmaceuticals. O site foi desenvolvido de acordo com os padrões legais e do setor para fornecer informações aos profissionais de saúde e ao público em geral sobre os tópicos de saúde da doença Alfamanosidose. A Chiesi Pharmaceuticals faz todos os esforços possíveis para incluir informações precisas e atuais. No entanto, as informações fornecidas neste site estão em constante atualização.


