Alpha Mannosidosis is a rare Lysosomal Storage Disease (LSD) caused by mutations affecting the alpha-mannosidase lysosomal enzyme. This enzyme is an exoglycosidase, that cleaves α-linked mannose residues of N-linked oligosaccharides. If the alpha mannosidase enzyme is impaired, degradation of glycoproteins is blocked and a progressive accumulation of mannose-rich oligosaccharides occurs in all tissues, leading to impaired cellular function and apoptosis.1

This impaired cellular function has a downstream impact upon organ systems, with typical presentation reflected as skeletal deformation, coarse facial features, hearing loss, cognitive disabilities, immune defects, and central nervous system involvement.2
The prevalence of Alpha Mannosidosis is not known exactly.
However, a number of reports from different countries estimate that it occurs in approximately one in every million babies born worldwide.3 The condition is often diagnosed and treated using a multi-disciplinary approach, involving paediatricians, orthopaedics, ophthalmologists, otologists neurologists, immunologists, neurosurgeons and physiotherapists.4
Alpha Mannosidosis is a progressive disorder, and its presence should be suspected in patients with mental cognitive disabilities, skeletal changes (eg swollen joints, curved spine), hearing loss and recurrent infections. Although children with the condition are often born seemingly normal, their condition deteriorates with age. Alpha Mannosidosis can impact on a patient’s quality of life in many ways, including their ability to live independently, socialise or find employment.5 6
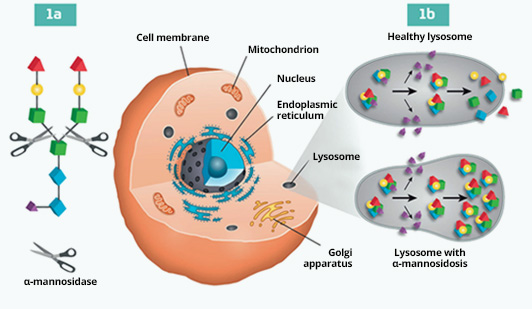
Mechanism of
the disease
The information on this website is intended only to provide knowledge of Alpha Mannosidosis disease health topics. This information should not be used in place of advice from your GP or other healthcare professional. If in doubt please contact your doctor for advice. This website has been produced by Chiesi Pharmaceuticals. The website has been developed in accordance with industry and legal standards to provide information for healthcare professionals and the general public about Alpha Mannosidosis disease health topics. Chiesi Pharmaceuticals makes every reasonable effort to include accurate and current information. However, the information provided in this website is not exhaustive.




