L’alpha-mannosidose est une maladie lysosomale rare due à des mutations de l’enzyme alpha-mannosidase lysosomale. Cette enzyme est une exoglycosidase qui sépare les résidus de l’alpha-mannose des oligosaccharides N-liés. Si l’enzyme alpha-mannosidase est endommagée, la dégradation des glycoprotéines est bloquée et des oligosaccharides riches en mannose s’accumulent progressivement dans tous les tissus, ce qui empêche le bon fonctionnement de la cellule et l’apoptose.1

Le dysfonctionnement cellulaire se manifeste par une déformation squelettique, des traits grossiers du visage, une perte auditive, un handicap cognitif, une défaillance immunitaire et des troubles du système nerveux central.2 La prévalence de l’ alpha-mannosidose n’est pas connue de manière précise. Toutefois, d’après un certain nombre de rapports de différents pays, il est estimé qu’elle touche environ un nouveau-né sur un million dans le monde.3 Cette maladie est souvent diagnostiquée et traitée selon une démarche multi-disciplinaire qui implique des pédiatres, orthopédistes, ophtalmologues, oto-rhino-laryngologues (ORL), neurologues, immunologues, neurochirurgiens et physiothérapeutes.4 L’alpha-mannosidose est une maladie progressive, et sa présence doit être suspectée chez les patients qui présentent un handicap cognitif mental, des changements squelettiques (ex. articulations enflées, colonne vertébrale courbée), une perte de l’audition et des infections récurrentes. Bien que les enfants atteints de la maladie semblent normaux à la naissance, leur état se détériore avec le temps. L’alpha-mannosidose peut influencer de différentes manières la qualité de vie des patients, comme par exemple leur capacité à vivre de manière indépendante, à se socialiser et à trouver un emploi.5 6
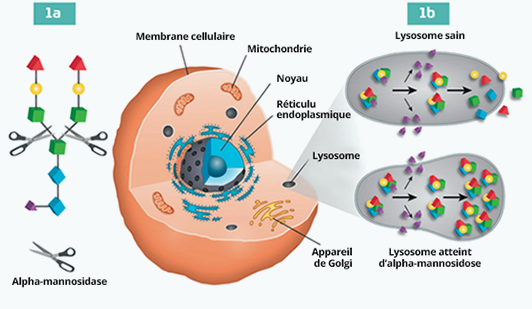
Le mécanisme de
la maladie
Les informations figurant sur ce site web sont uniquement destinées à fournir des connaissances sur les thématiques de santé liées à l’alpha-mannosidose. Ces informations ne doivent pas remplacer les conseils de votre médecin ou d’un autre professionnel de santé. En cas de doute, veuillez contacter votre médecin pour obtenir des conseils. Ce site web a été réalisé par Chiesi Pharmaceuticals. Il a été développé conformément aux normes industrielles et juridiques afin de fournir des informations aux professionnels de santé et au grand public sur des thématiques de santé liées à l’alpha-mannosidose. Chiesi Pharmaceuticals fait tout son possible pour inclure des informations précises et actuelles. Toutefois, les informations fournies sur ce site ne sont pas exhaustives.

