α-Mannosidose ist eine seltene Lysosomale Speicherkrankheit (LSK oder aus dem Englischen LSD), die durch Mutationen in dem Gen verursacht wird, das das Enzym α-Mannosidase kodiert. Dieses Enzym ist eine Exoglykosidase, die α-gebundene Mannosereste von N-gebundenen Oligosacchariden abspaltet. Wenn das Enzym α-Mannosidase beschädigt ist, wird der Abbau der Glykoproteine blockiert, und es kommt in allen Geweben zu einer fortschreitenden Anhäufung von Mannose-reichen Oligosacchariden, was zu einer Beeinträchtigung der Zellfunktion und zur Apoptose führt.1

Diese gestörte Zellfunktion wirkt sich dann auf verschiedene Organsysteme aus, und es entsteht das typische Erscheinungsbild mit Skelettdeformationen, groben Gesichtszügen, Hörverlust, kognitiven Störungen, Immundefekten und einer Beteiligung des zentralen Nervensystems.2 Die Prävalenz der α-Mannosidose ist nicht genau bekannt. In einer Reihe von Berichten aus verschiedenen Ländern wird jedoch geschätzt, dass die Erkrankung weltweit bei etwa einem von einer Million Neugeborenen auftritt.3 Sie wird häufig mit einem multidisziplinären Ansatz diagnostiziert und behandelt, an dem Kinderärzte, Orthopäden, Augenärzte, Ohrenärzte, Neurologen, Immunologen, Neurochirurgen und Physiotherapeuten beteiligt sind.4 α-Mannosidose ist eine fortschreitende Erkrankung, und sollte bei Patienten und Patientinnen mit geistig-kognitiven Störungen, Skelettveränderungen (z. B. geschwollene Gelenke, gekrümmte Wirbelsäule), Hörverlust und wiederkehrenden Infektionen in Betracht gezogen werden. Obwohl Kinder mit dieser Erkrankung oft scheinbar normal geboren werden, verschlechtert sich ihr Zustand dann mit zunehmendem Alter. α-Mannosidose kann sich in vielerlei Hinsicht auf die Lebensqualität der Patienten und Patientinnen auswirken, unter anderem in der Fähigkeit, unabhängig zu leben, soziale Kontakte zu knüpfen oder eine Arbeitsstelle zu finden.5, 6
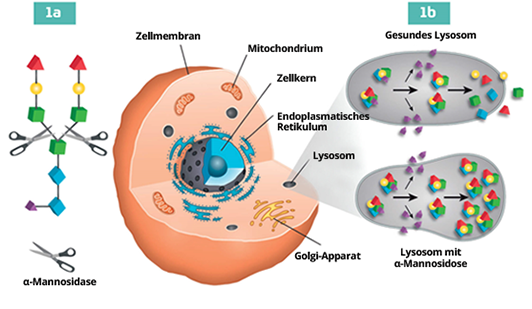
Krankheitsmechanismus
Die Informationen auf dieser Website sollen lediglich Wissen über die mit dem Krankheitsbild α-Mannosidose verbundenen Gesundheitsaspekte vermitteln. Die gegebenen Informationen sollen nicht die Beratung durch Ihren Hausarzt oder andere medizinische Fachkräfte ersetzen. Falls Zweifel auftreten sollten, wenden Sie sich bitte an Ihren Arzt und lassen Sie sich von ihm beraten. Diese Website wurde von Chiesi Pharmaceuticals erstellt. Die Website wurde in Übereinstimmung mit den für die Branche geltenden und den gesetzlichen Bestimmungen entwickelt, um das medizinische Fachpersonal und die allgemeine Öffentlichkeit über verschiedene Gesundheitsaspekte, die mit der Erkrankung α-Mannosidose in Verbindung stehen, zu informieren. Chiesi Pharmaceuticals versucht, Ihnen immer möglichst genaue und aktuelle Informationen bereitzustellen. Die auf dieser Website enthaltenen Informationen erheben jedoch nicht den Anspruch auf Vollständigkeit.


