Hier finden Sie Antworten auf die gängigsten Fragen zur α-Mannosidose.
Die Fragen sind nach Themenkreisen geordnet, um Ihnen die Suche zu erleichtern.
Bewegen Sie Ihre Maus auf die einzelnen Fragen, um die gesuchte Antwort zu finden.
Die α-Mannosidose ist eine seltene Erbkrankheit, die bei Kindern und Erwachsenen zu Skelettdeformationen, groben Gesichtszügen, Hörverlust, mentaler Retardierung, Problemen mit dem Immunsystem, psychiatrischen Störungen und Verhaltensauffälligkeiten führen kann.1 Wenn das Gen, das die Informationen für die Synthese der α-Mannosidase kodiert, nicht einwandfrei funktioniert, wird das Enzym nicht richtig hergestellt und es entsteht eine α-Mannosidose.2 Dieses Enzym ist in Zellkompartimenten aktiv, die als Lysosomen bezeichnet werden. Die bauen verschiedene Materialien ab und wiederverwerten sie. In den Lysosomen hilft das Enzym dabei, langkettige Zuckermoleküle (die sog. Oligosaccharide) zu spalten. Oligosaccharide werden für den Aufbau von Knochen, Knorpel, Haut, Sehnen und vielen anderen Geweben im Körper verwendet.3, 4 Bei Menschen mit einer α-Mannosidose bleiben die teilweise abgebauten Zucker im Körper zurück und häufen sich mit der Zeit an. Dadurch werden die Zellen zunehmend geschädigt.5Säuglinge zeigen häufig nur wenige Anzeichen der Erkrankung. Wenn jedoch immer mehr Zellen durch die Anhäufung von Oligosacchariden geschädigt werden, treten dann auch die entsprechenden Symptome auf.6
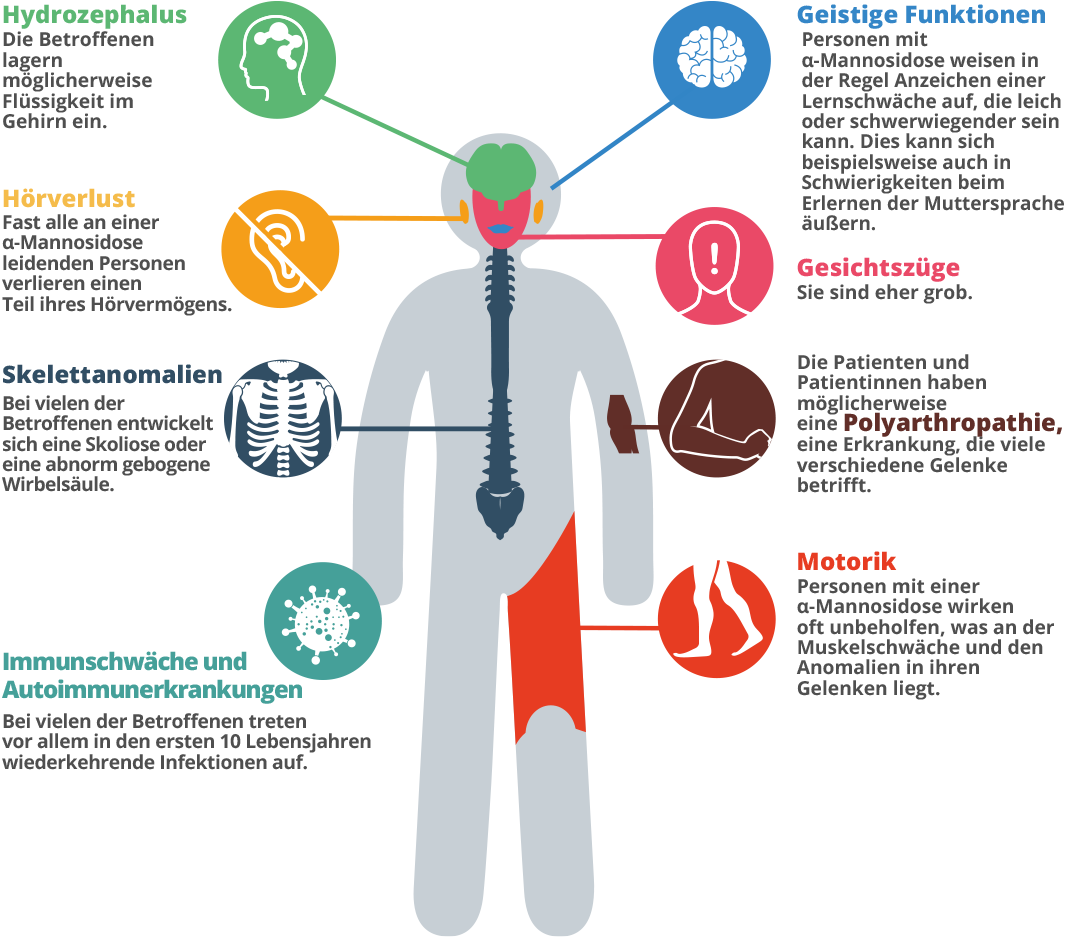
Die Symptome der α-Mannosidose können, wie bei anderen verwandten Erkrankungen auch, sehr unterschiedlich sein.7 Es handelt sich um eine fortschreitende Krankheit, die sich im Laufe der Zeit auf unterschiedliche Weise äußert.8 Da sie eine so große Bandbreite an Symptomen aufweist und jeder Mensch sein eigenes, einzigartiges Muster an Symptomen entwickelt, kann es lange dauern, bis die Diagnose gestellt ist.9 Während des ersten Lebensjahrzehnts kann ein Kind mit dieser Krankheit häufige Infektionen, Hörprobleme, charakteristische Gesichtszüge und Entwicklungsverzögerungen aufweisen.10, 11 Die Eltern stellen unter Umständen fest, dass ihr Kind eine Art Muskelschwäche hat.12 Möglicherweise liegt auch ein ungewöhnliches Körpermerkmal, wie z. B. ein Klumpfuß, ein großer Kopf, ein ungewöhnliches Aussehen oder vielleicht ein gebeugter Rücken, vor. Ein Kind hat möglicherweise Aufmerksamkeitsprobleme und Schwierigkeiten mit dem Hören.13, 14 Ein Symptom kann evtl. für sich stehen, eine Kombination der Symptome zur gleichen Zeit ist jedoch kein Zufall mehr. Betrachten Sie alle Symptome und lassen Sie sich von Ihrem Hausarzt an einen Stoffwechselspezialisten überweisen. In den 20er und 30er Jahren kann ein Erwachsener unter Knochenproblemen und Bewegungsschwierigkeiten leiden, hierzu gehören unter anderem z. B. Gelenkprobleme, Schwellungen, ein unsicherer Gang und Muskelschwäche. Später können die Patienten und Patientinnen dann Rollstuhl-pflichtig werden, da sie nicht mehr alleine gehen können.15 Es können Verhaltens- oder psychiatrische Störungen auftreten, die sich in Form von Verwirrungszuständen äußern können, die gelegentlich mit Ängsten, Depressionen oder Halluzinationen kombiniert sind.16 Ein unabhängiges Leben wird schwierig sein, und Patienten und Patientinnen mit α-Mannosidose werden möglicherweise sozial isoliert.17, 18 Die langfristige Prognose ist ungünstig.19
Es wurden drei klinische Untertypen vorgeschlagen:37
Angesichts der Vielfalt der dokumentierten Mutationen, des breiten Spektrums und des Schweregrades der Symptome sowie der fehlenden Verbindung zwischen bestimmten Mutationen und dem klinischen Untertyp wird die Krankheit klinisch als ein Kontinuum betrachtet, das von leicht bis schwer reicht.38, 39
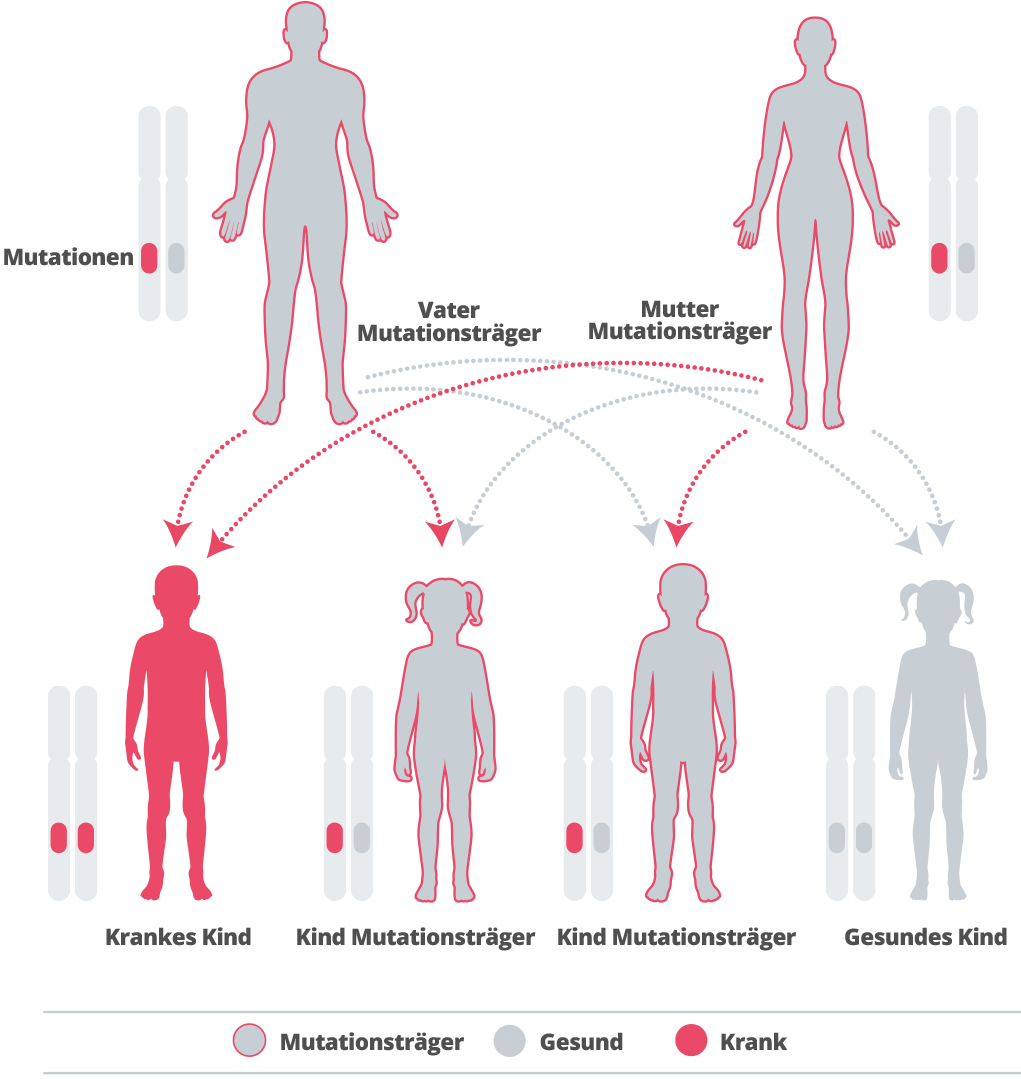
α-Mannosidose wird vererbt, d. h. sie ist familiär bedingt.40 Wir erben die Gene von unseren Eltern. Und zwar erben wir je eine Kopie aller Gene von jedem Elternteil. Einige Gene, die wir erben, sind „rezessiv“, was bedeutet, dass beide Kopien dieses Gens vererbt werden müssen, damit es sich auf unsere Entwicklung auswirken kann.41, 42 Die α-Mannosidose wird von einem rezessiven Gen verursacht.43Wenn ein Erwachsener, der das veränderte Gen trägt, einen Partner hat, der ebenfalls Träger ist, besteht bei jeder Schwangerschaft eine Wahrscheinlichkeit von 25 %, dass das Kind das defekte Gen von jedem Elternteil erbt und an der Krankheit leidet. Die Wahrscheinlichkeit, dass nicht erkrankte Geschwister von an α-Mannosidose erkrankten Personen Überträger sind, besteht in zwei von drei Fällen.44, 45 Da die Krankheit so selten ist, ist die Wahrscheinlichkeit, einen Partner zu haben, der auch Überträger ist, sehr gering, es sei denn, die Betroffenen gehören zur selben Familie.46 Wenn ein Paar bereits ein Kind mit der Krankheit hat, wird eine genetische Beratung empfohlen, um herauszufinden, wie hoch die Wahrscheinlichkeit ist, dass die Krankheit auch bei zukünftigen Nachkommen auftritt.47
Die Prävalenz der α-Mannosidose ist nicht genau bekannt. In einer Reihe von Berichten aus verschiedenen Ländern wird jedoch davon ausgegangen, dass sie weltweit bei etwa einem von einer Million Neugeborenen vorkommt.48
Ihr Arzt kann mit einer Reihe von sehr einfachen Tests feststellen, ob eine α-Mannosidose vorliegt, hierzu gehören unter anderem:
Es gibt auf der ganzen Welt Selbsthilfegruppen, die gerne mit Ihnen ihre Informationsquellen teilen, Sie verstehen und Ihnen mit Rat und Tat zur Seite stehen. Mit wenigen Klicks können Sie Kontakt mit zahlreichen Organisationen und Selbsthilfegruppen aufnehmen. Patientenorganisationen:
 ist die internationale Selbsthilfegruppe für α-Mannosidose. ist die Selbsthilfegruppe für Familien in Neuseeland.. ist ein Netzwerk von Gesundheitsdienstleistern und Patientengruppen, das in ganz Europa zu finden ist und Menschen mit erblichen Stoffwechselstörungen wie der α-Mannosidose unterstützt.
ist die internationale Selbsthilfegruppe für α-Mannosidose. ist die Selbsthilfegruppe für Familien in Neuseeland.. ist ein Netzwerk von Gesundheitsdienstleistern und Patientengruppen, das in ganz Europa zu finden ist und Menschen mit erblichen Stoffwechselstörungen wie der α-Mannosidose unterstützt.Wenn eine α-Mannosidose diagnostiziert wird, kann das erhebliche emotionale Auswirkungen auf die Patienten und Patientinnen und deren Betreuer haben. Es müssen sowohl die primäre als auch die spezifische Versorgung und der Zugriff auf soziale Dienstleistungen sichergestellt sein. Darüber hinaus sollten die ernährungswissenschaftliche und psychologische Unterstützung regelmäßig bewertet und überdacht werden.53 Die klinische Versorgung sollte proaktiv erfolgen, wobei die Ärzte entsprechende Pläne aufstellen sollten, um die Folgen der Erkrankung und etwaige Komplikationen für die Patienten und Patientinnen möglichst zu begrenzen.54 Chirurgische Eingriffe an den Gelenken und anderen Teilen des Skeletts sollten ggf. in Betracht gezogen werden.55 Für weitere Informationen zum Umgang mit der Erkrankung und den Behandlungsmöglichkeiten sprechen Sie bitte mit Ihrem Arzt.
Die Informationen auf dieser Website sollen lediglich Wissen über die mit dem Krankheitsbild α-Mannosidose verbundenen Gesundheitsaspekte vermitteln. Die gegebenen Informationen sollen nicht die Beratung durch Ihren Hausarzt oder andere medizinische Fachkräfte ersetzen. Falls Zweifel auftreten sollten, wenden Sie sich bitte an Ihren Arzt und lassen Sie sich von ihm beraten. Diese Website wurde von Chiesi Pharmaceuticals erstellt. Die Website wurde in Übereinstimmung mit den für die Branche geltenden und den gesetzlichen Bestimmungen entwickelt, um das medizinische Fachpersonal und die allgemeine Öffentlichkeit über verschiedene Gesundheitsaspekte, die mit der Erkrankung α-Mannosidose in Verbindung stehen, zu informieren. Chiesi Pharmaceuticals versucht, Ihnen immer möglichst genaue und aktuelle Informationen bereitzustellen. Die auf dieser Website enthaltenen Informationen erheben jedoch nicht den Anspruch auf Vollständigkeit.


