Найдите ответы на самые частые вопросы об альфа-маннозидозе.
Вопросы структурированы по темам для вашего удобства
Наведите курсор на каждый вопрос, чтобы найти нужный вам ответ.
Альфа-маннозидоз — это редкое наследственное заболевание, которое вызывает развитие аномалий скелета, грубых черт лица, потерю слуха, отставание в умственном развитии, проблемы с иммунной системой, психические нарушения, поведенческие проблемы у детей и взрослых с данной патологией 1.
Если ген, который отвечает за создание альфа-маннозидазы, работает неправильно, данный фермент также образуется неправильно, что приводит к развитию альфа-маннозидоза 2.
Этот фермент работает в лизосомах — органеллах, которые переваривают и перерабатывают материалы внутри клетки Внутри лизосом данный фермент помогает разрушать длинные цепи молекул сахаров (олигосахаридов) Олигосахариды используются для построения костей, хрящей, кожи, сухожилий и многих других тканей в организме человека 3, 4.
У людей с альфа-маннозидозом частично расщепленные сахара остаются в организме и накапливаются с течением времени. Это приводит к прогрессирующему повреждению клеток 5. У новорожденных детей симптомы заболевания практически отсутствуют и начинают появляться только по мере того, как все больше и больше клеток повреждаются вследствие накопления в них сахаров 6.
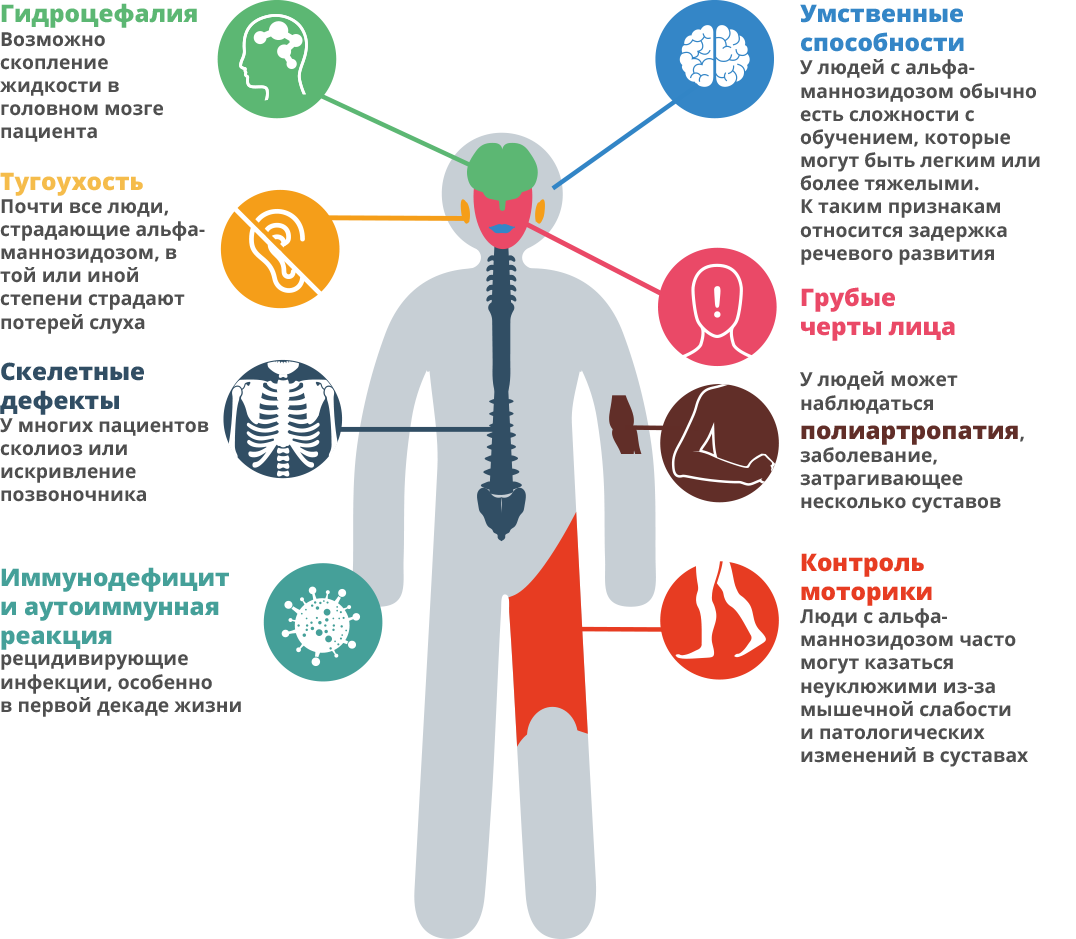
Симптомы альфа-маннозидоза, как и многих похожих заболеваний, могут в значительной степени варьировать 7.
Это прогрессирующее заболевание, которое проявляется по-разному в различные моменты времени 8. Поскольку спектр возможных симптомов достаточно широк, у каждого пациента отмечается свой уникальный набор симптомов, что затрудняет быструю постановку диагноза 9.
В течение первых 10 лет жизни у детей могут иметь место частые инфекции, нарушения слуха, грубые черты лица, задержка развития 10, 11.
Родители могут заметить некоторую мышечную слабость у ребенка 12. Могут отмечаться особенности строения скелета, например косолапость, большой размер головы, согнутая спина.У ребенка могут быть проблемы со вниманием и проблемы со слухом 13, 14. Отдельные симптомы могут наблюдаться у здоровых детей, но если они присутствуют все вместе — это уже не просто совпадение.
Проанализируйте все имеющиеся симптомы и обратитесь за консультацией к специалисту по заболеваниям обмена веществ, попросив вашего семейного врача направить вас к нему.
В возрасте от 20 до 40 лет взрослый человек может испытывать проблемы с костями и трудности с передвижением, в частности появляются проблемы с суставами, отеки, неустойчивая походка и мышечная слабость.В конечном счете, пациенты начинают зависеть от других людей и могут перемещаться только в инвалидной коляске, так как больше не могут ходить самостоятельно 15. Могут возникнуть поведенческие или психические проблемы, которые могут проявляться в виде эпизодов спутанности сознания, тревожности, депрессии или развития галлюцинаций 16. Пациенты с альфа-маннозидозом часто не могут жить без посторонней помощи, и они могут стать изолированными от общества 17, 18. Долгосрочный прогноз при данном заболевании неблагоприятный 19.
Выделяют следующие клинические разновидности заболевания.37
Однако с учетом различных мутаций, широкого спектра мутаций и их выраженности, а также отсутствия корреляций между определенными мутациями и клинической картиной клинически заболевание может варьировать от легкой до тяжелой степени 38 39
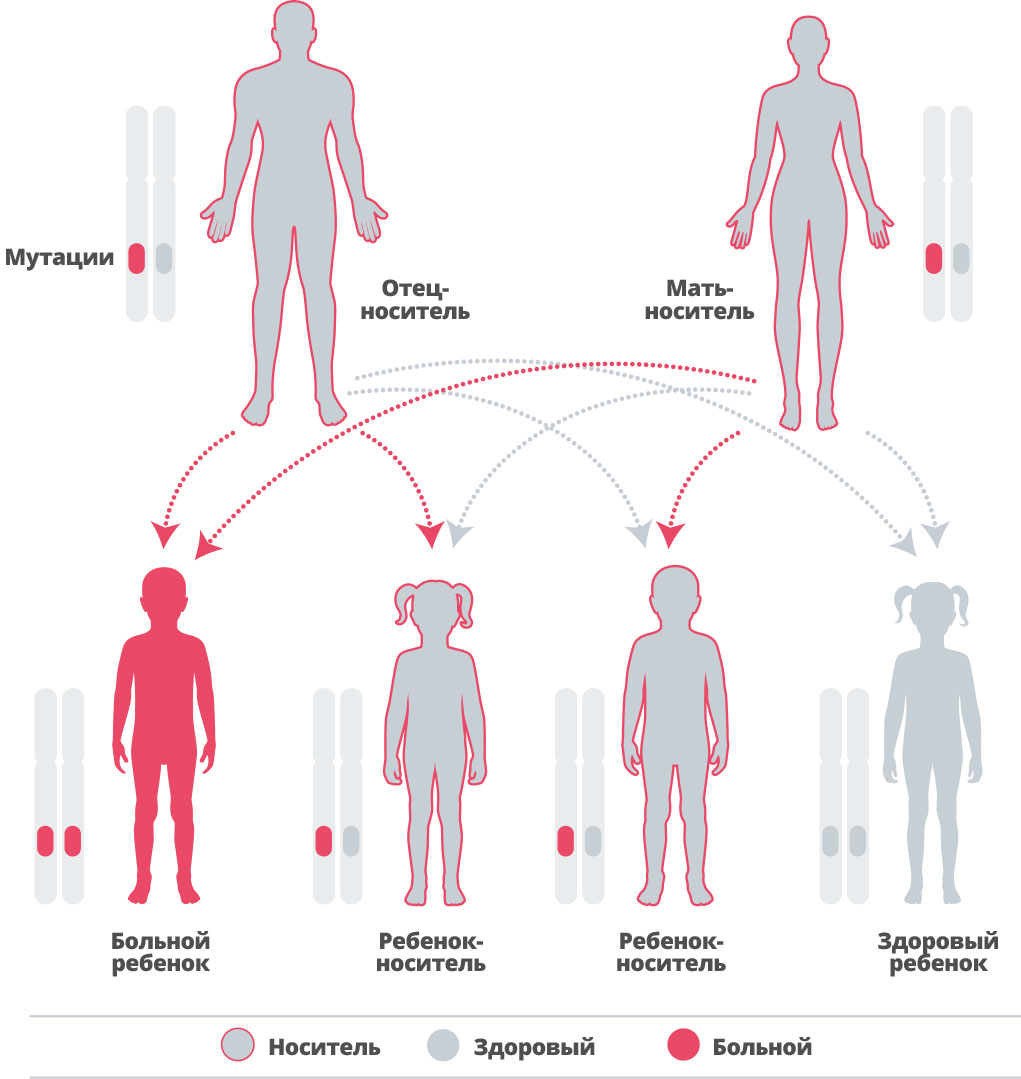
Альфа-маннозидоз является наследственным заболеванием, то есть передается внутри семьи 40. Гены мы наследуем от своих родителей. Мы наследуем по копии генов от каждого из родителей.
Некоторые из наследуемых нами генов являются «рецессивными». Это значит, что только оба гена, унаследованные вместе, могут повлиять на наше развитие 41, 42. Развитие альфа-маннозидоза связано с рецессивным геном 43.
При каждой беременности у пары, в которой оба партнера гетерозиготны по болезнетворной мутации гена MAN2B1, вероятность рождения больного ребенка составляет 25 %, вероятность рождения здорового ребенка-носителя — 50 % и вероятность рождения здорового ребенка, который не является носителем, — 25 %.44
Поскольку заболевание очень редкое, вероятность встретить партнера, который тоже является носителем такой мутации, мала, если эти люди не являются членами одной семьи 45. Если у пары уже есть ребенок с такой патологией, им необходимо пройти генетическое обследование, которое позволит установить вероятность развития заболевания у их будущих детей46.
Точная частота альфа-маннозидоза неизвестна.Однако, согласно имеющейся информации относительно его распространенности в различных странах мира, данное заболевание встречается с частотой один случай на 1 миллион рожденных детей по всему миру.47
Ваш врач может назначить ряд очень простых анализов на альфа-маннозидоз, например:
Существует сообщество, которое готово предоставить вам необходимую информацию, оказать поддержку и дать совет. Существует ряд организаций и групп поддержки, которые находятся от вас на расстоянии щелчка мышью.
Пациентские организации:
Диагноз альфа-маннозидоза может в значительной степени повлиять на эмоциональное состояние пациентов и лиц, ухаживающих за ними.
Необходимо обеспечить пациентам доступ к первичной и специализированной медицинской помощи и социальным услугам, а также постоянной психологической поддержке 52. Медицинская помощь должна носить упреждающий характер для профилактики развития осложнений и негативных последствий данного заболевания 53. Возможно, потребуется хирургическое ортопедическое вмешательство54. Для получения более подробной информации о вариантах лечения, пожалуйста, обратитесь к вашему лечащему врачу.
Информация на этом веб-сайте предоставлена в целях ознакомления со свойствами заболевания альфа-маннозидоз. Эта информация не заменяет рекомендации лечащего врача.
Этот веб-сайт создан компанией Кьези Фармацевтичи С.п.А. Веб-сайт разработан в соответствии с отраслевыми и правовыми стандартами для предоставления медицинским работникам и широкой общественности информации о проблемах со здоровьем, связанных с заболеванием альфа-маннозидоз. Компания Кьези Фармацевтичи С.п.А. прилагает все разумные усилия для добавления точной и актуальной информации, однако информация, представленная на этом веб-сайте, не является исчерпывающей.


