This rare disease was described for the first time by Swedish Physician Okerman in 1967, and is one of a wide range of Lysosomal Storage Disorders, caused by mutations affecting the alpha-mannosidase lysosomal enzyme, resulting in its deficiency. This enzyme is an exoglycosidase, that cleaves α-linked mannose residues of N-linked oligosaccharides.3
Figure 1a. α-mannosidase cleaves the alpha-linked mannose residues of N-linked oligosaccharides.

Figure 1b. In healthy cells, α-mannosidase in the lysosomes acts in the sequential degradation of complex glycoproteins. Smaller breakdown products leave the lysosome. In α-mannosidosis accumulation of α-mannosyl rich N-linked oligosaccharides leads to lysosomal engorgement and disruption of normal cell function.
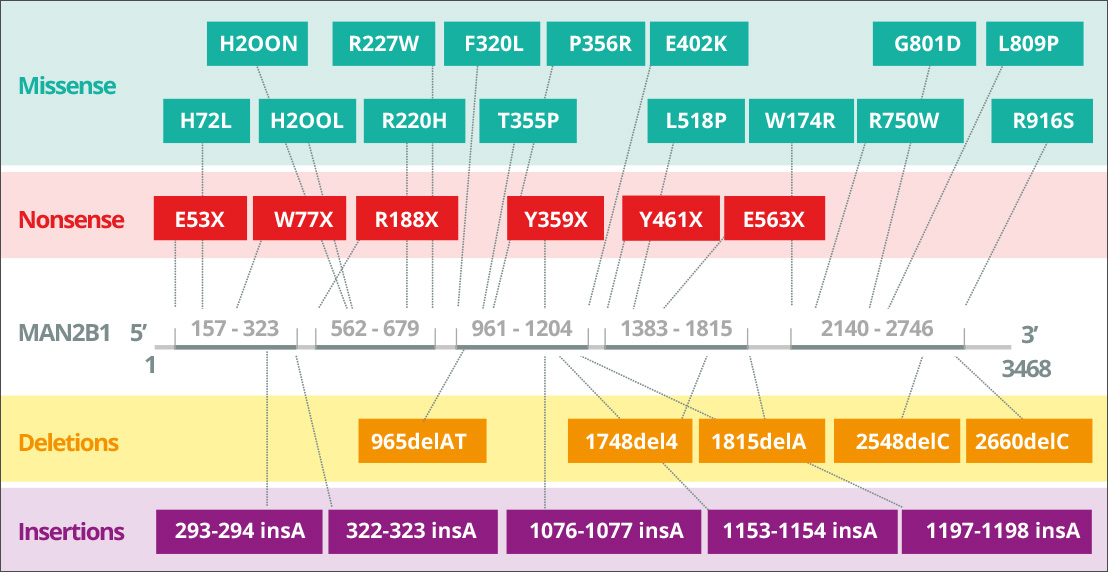
Alpha mannosidosis is caused by hereditary mutations in the MAN2B1 (LAMAN) gene encoding lysosomal α-mannosidase4A. Alpha mannosidosis has autosomal recessive inheritance. The MAN2B1 gene is composed of 24 exons and encodes a 1011 amino acid polypeptide that is post-translationally modified in the endoplasmic reticulum5A. During maturation and endosomal transport of MAN2B1 to the lysosomes it is proteolytically cleaved into three major polypeptides named “abc”, “d” and “e” of 70, 42 and 15 kDa, respectively6. Further specific, processing of the 70 kDa subunit results in a total of five different polypeptides. The level of MAN2B1 expression appears to be highest in lung, kidney, pancreas and peripheral blood leukocytes7A. In the CNS, the highest level of expression appears to be in corpus callosum and spinal cord, whereas considerably lower levels are observed in the larger structures, which include cerebellum, cerebral cortex, frontal and temporal lobes. However, the significance (if any) of such variations is not clear at present7A.
Mutations in MAN2B1 lead to loss of lysosomal alpha mannosidase activity8A. Depending on the causative MAN2B1 mutation, mutant MAN2B1 proteins have been detected in subcellular compartments such as endoplasmic reticulum and lysosomes9A. For instance, the protein can be folded incorrectly and arrested in the endoplasmic reticulum, or it can be folded correctly and transported to the lysosomes in an inactive form10A. To date, 155 variants from 191 patients have been identified and partly characterised at the biochemical level11A.
If the alpha mannosidase enzyme is impaired, there is a reduction in the degradation of glycoproteins and a progressive accumulation of mannose-rich oligosaccharides in all tissues, leading to impaired cellular function and apoptosis.13
Alpha Mannosidosis is inherited in an autosomal recessive fashion, caused by mutations in the gene MAN2B1, located on chromosome 19.
The phenotypic variability is high, even between siblings with identical genotypes14. In addition to genetic factors, environmental factors may influence the disease. For instance, exposure to pathogens may cause recurrent infections and a worsening of disease symptoms.15
The information on this website is intended only to provide knowledge of Alpha Mannosidosis disease health topics. This information should not be used in place of advice from your GP or other healthcare professional. If in doubt please contact your doctor for advice. This website has been produced by Chiesi Pharmaceuticals. The website has been developed in accordance with industry and legal standards to provide information for healthcare professionals and the general public about Alpha Mannosidosis disease health topics. Chiesi Pharmaceuticals makes every reasonable effort to include accurate and current information. However, the information provided in this website is not exhaustive.


